
Overview
This notebook demonstrates an end-to-end gene expression analysis workflow starting from a raw GEO Series Matrix file. The focus is on a core real-world bioinformatics skill: turning “messy” public repository files into analysis-ready matrices and metadata, then performing basic differential expression and exploratory analysis.
Although the dataset is public and compact, the same workflow scales to large transcriptomics studies and is directly applicable in oncology pipelines.
- Links: GitHub
- Status: Complete
Why this project
This project demonstrates a reproducible end-to-end gene expression analysis workflow turning chaotic public repository data into analysis-ready matrices and metadata.
Highlights
- Parsing GEO Series Matrix structure:
- detecting !series_matrix_table_begin / !series_matrix_table_end
- separating metadata lines from expression table
- Building a clean expression matrix:
- rows = genes
- columns = samples
- Extracting phenotype labels (Tumor vs Normal) from sample characteristics
Results
| Figure |
|---|
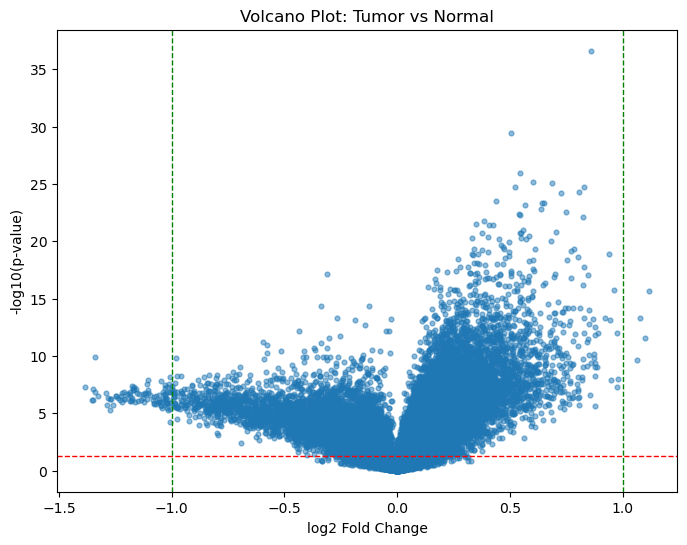 Figure 1: Volcano plot Tumor vs Normal |
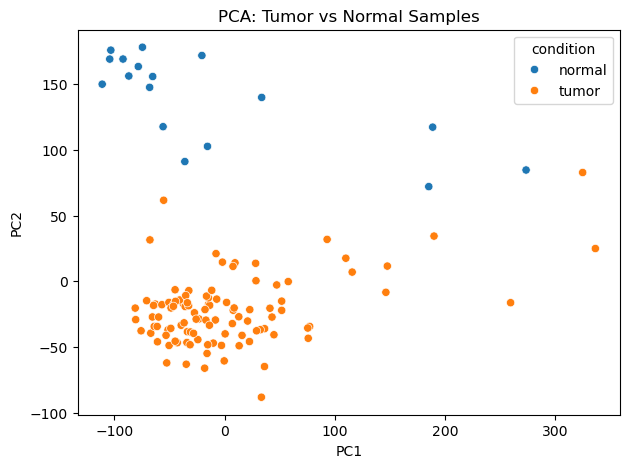 Figure 2: PCA plot Tumor vs Normal |
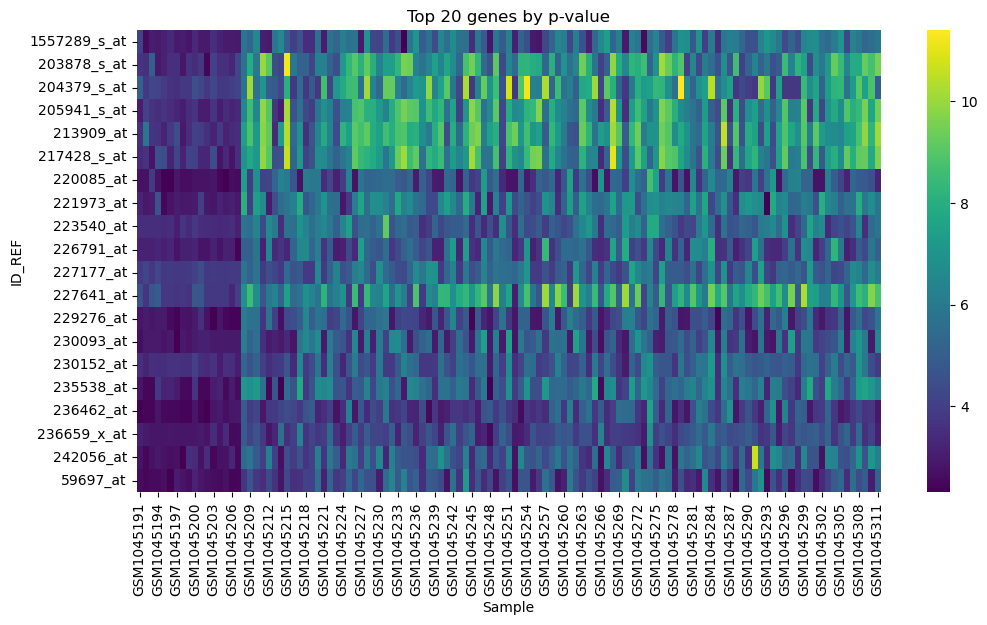 Figure 3: Top 20 genes based on p-value |
How to run
git clone https://github.com/Anwesha19-prog/Breast-Cancer-Microarray-Analysis
cd Breast-Cancer-Microarray-Analysis
pip install pandas numpy scipy matplotlib seaborn scikit-learn
jupyter notebook Project-1_clean.ipynb